index
.
MassKinetics Tutorial
1. Introduction
Here we will show you in detail how to use the MassKinetics program in a simple case. In this tutorial various MassKinetics window names and menu items are printed red italic bold; parameters to be selected by underlined bold characters. Detailed description of the theory, terminology, parameters, approximations used in MassKinetics can be found in our recent review (L. Drahos and K. Vékey, J. Mass Spectrom. 36, 237 (2001)).
We take the example published few years ago in a tutorial article in JMS on ion energetics (K. Vékey, J. Mass Spectrom. 31, 445-463 (1996)) – this is also a good place to start understanding fundamentals of mass spectrometry. First we show you how to set up a given experiment, next we show you how you can calculate various features for this model reaction. The ‘Tips and Tricks’ section of our homepage gives you ideas how to use MassKinetics for more advanced calculations. Tips for application of MassKinetics to chemical problems can be found in the ‘Examples’ and ‘References’ sections of this homepage.
Please note that we are in the process of developing and testing the program, and there are likely to be many errors and bugs in it. If you find one, please inform us, preferably sending us the project file ('mkp' extension) and a short explanation of the bug.
First, you have to start the program (by double clicking on its icon) and accept the license agreement. After that you get to the main window of MassKinetics:
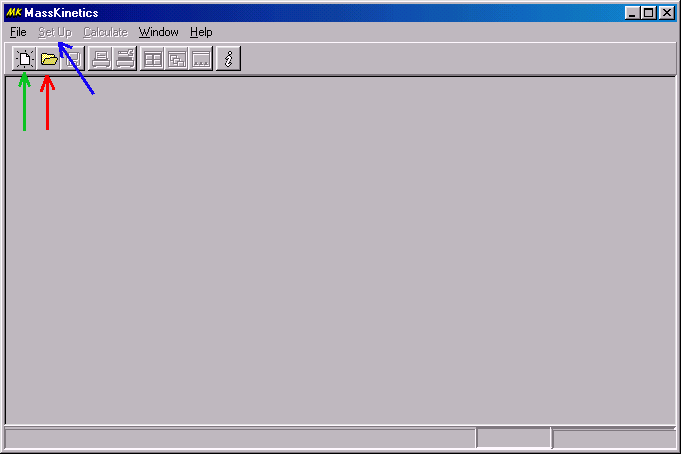
If it is your first application, you have to create a new project (green arrow), otherwise you may open an existing one (red arrow). You may save your MassKinetics project any time, and you may change its name by "save as". In general, if you want to select or modify an item in the dialog box you have to click or double click on it – then you will either be able to type in the proper value, or you will be led to a new dialog box which you have to fill out properly.
If you want to learn, how to create a new MassKinetics project, continue with the next section. If you want to learn, how to modify an existing project and use it for various model calculations, go to the Tips & Tricks section.
2. Create a new project: CID fragmentation (buthyl radical and buthene loss) of buthylbenzene
After you create a new project (green arrow in the previous diagram) you will be able to click on the Setup menu (blue arrow) to define your experiment. This brings up a dialog box and takes you to the "Comments" tab of this dialog:
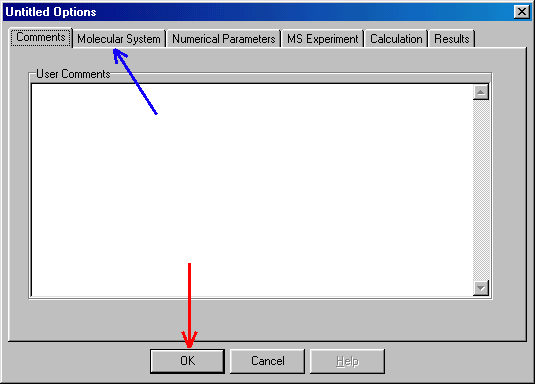
In the Comments window you can type (now, or any time later) comments about your MassKinetics project. To specify/modify the MassKinetics project, you have to go through Molecular System, Numerical Parameters, MS Experiment, Calculation and Results tabs. You can do it in any sequence you prefer, and you can return to a previous one at will. You may save the setup file at any point. To do so you have to click OK button (red arrow), than in the file menu click "save" or "save as". In fact, it is advisable to save the setup as you progress, possibly using different names (e.g. Test1, Test2 etc.). Clicking on the Molecular System (blue arrow) you get to the "General" section of the most complex page:
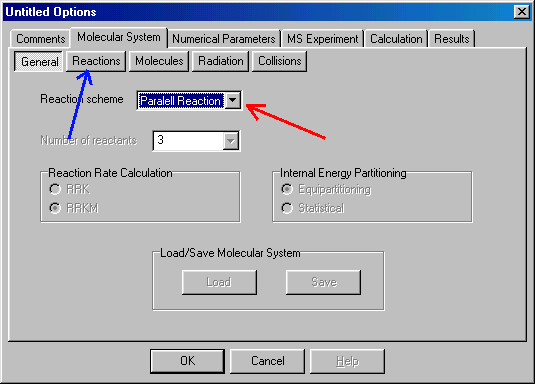
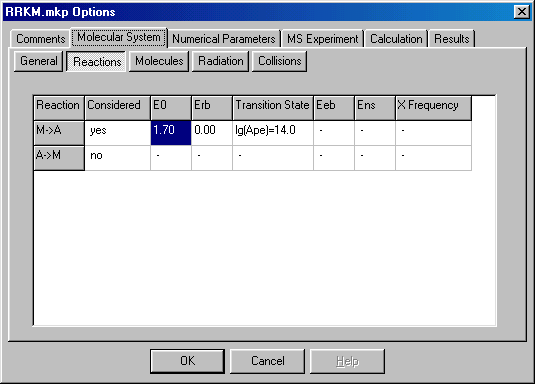
Here you select (red arrow) the required reaction type ("parallel reaction"). In future versions of MassKinetics you will have several more options, now you proceed to the Reactions tab (blue arrow).
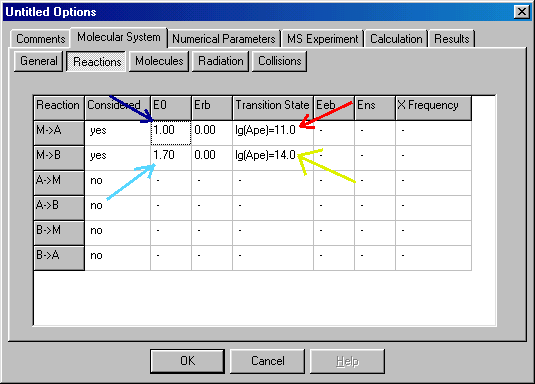
Here you define critical (~ activation) energy (E0, given in eV units) and the transition state for each reaction channel. To specify or modify values (here and elsewhere) you have to click or double click on them. After that you can either type in the correct value, or a new dialog box will appear, which you can fill out. Click on the critical energy of the first reaction (corresponding to propene elimination, blue arrow), and type in 1.0 (E0 in eV units). E0 of the second process (prolyl radical loss, 1.7 eV) is filled out similarly (light blue arrow). To specify the transition states you have to double-click on them (red and yellow arrows), than another dialog box appears:
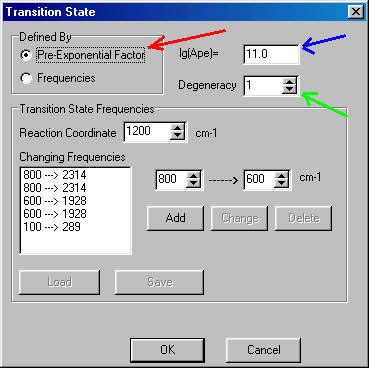
You can characterize the Transition State window using the pre-exponential (or frequency) factor. To do so, select the Pre-Exponential Factor (red arrow), and fill out the lg(Ape) value (the logarithm of pre-exponential factor, blue arrow). For propene elimination it is 11.0, for propyl radical elimination it is 14.0. A more accurate alternative is using transition state frequencies – see ‘Tips and Tricks’ how to do that. Beside the pre-exponential factor you have to define reaction degeneracy as well (green arrow) – in the example it is 1 (the default value) for both reactions. When you finished defining the transition state click OK button to accept it, than you return to the Reactions section. When you have defined both transition states you select the Molecules tab:
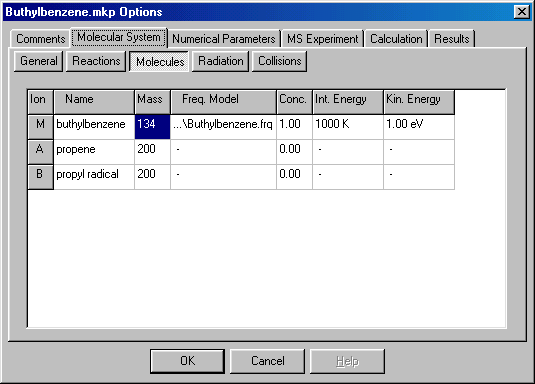
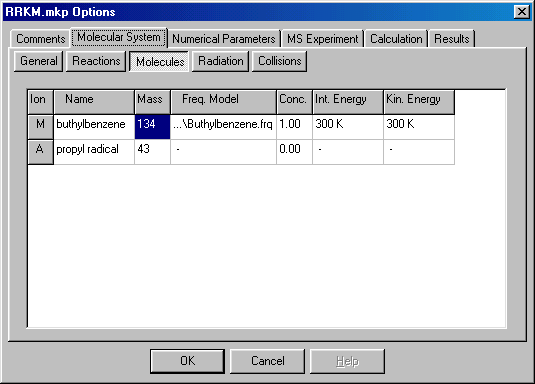
In the table here you specify the molecular parameters and initial state of the compounds. The first column (red arrow) gives the name of the ion – it is only for your information, and you can rename them as you wish by clicking on it and typing the new name. Rename Fragment A as ‘propene’, Fragment B as ‘propyl radical’. The second column (blue arrow) gives the molecular mass. For the precursor (molecular) ion you have to specify it (in Daltons, for buthylbenzene it is 134), for the fragments it is irrelevant. The third column (green arrow) defines the Frequency Model – a list of molecular normal frequencies (used in RRKM calculations), necessary only for the precursor ion. Double clicking on it will give you a new dialog box:
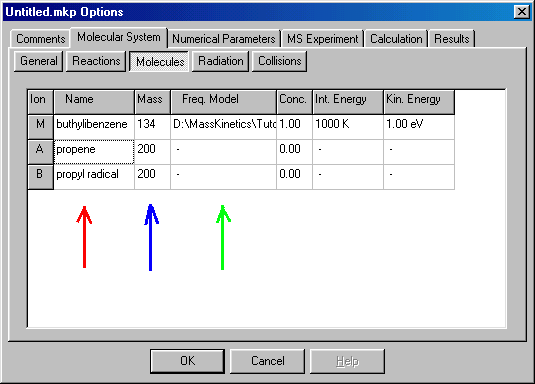
In the "Set Frequency Model" dialog box you can specify the frequency model by clicking on the "Load Frequency File" button (red arrow), which prompts you to select the required frequency file:
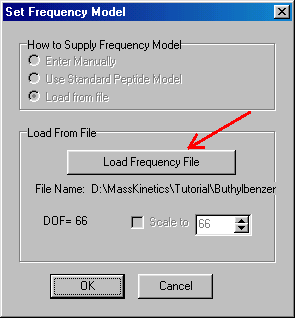
The MassKinetics Demo contains the frequency file of buthylbenzene (Buthylbenzene.frq) in the MassKinetics folder, for other compounds you will have to prepare that in advance. The frequency file is text file (you can create it using any text editor program) containing all molecular normal frequencies separated by Enter. Note that this file has to have an .frq or txt extension Note that buthylbenzene has altogether 66 (3N-6) molecular frequencies. After selecting the frequency file you click "Open" Button, which takes you back to the "Set Frequency Model" box, where you can accept the frequency file clicking the OK button, which takes you back to the Molecules window:
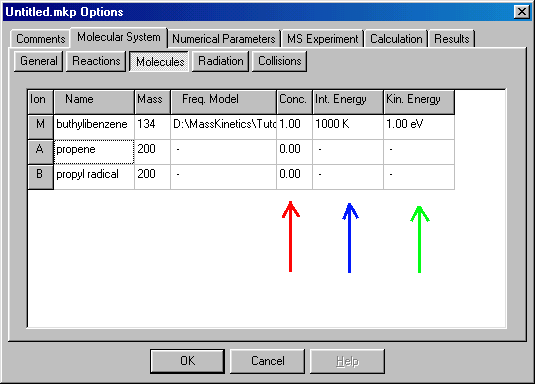
The next column (red arrow) here is the initial concentration (Conc.) – at the time of ion formation the initial concentration of the molecular ion is unity, while it is zero for all other species (these are the default values). The Int. Energy column (blue arrow) specifies the initial internal energy (necessary to specify only for the precursor). When you double click on it a new window appears:
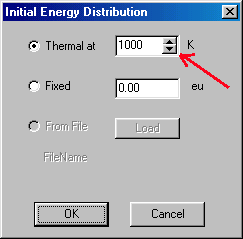
In the Internal Energy Distribution window you can either specify a fixed, single value, or a thermal distribution. The latter is often a good approximation – the mean internal energy is defined using certain temperature. For buthylbenzene in EI ionization you may define internal energy distribution to correspond to 1000 K. You click on "Thermal at:" than type in the temperature (1000) which is given in K (red arrows). Accept it by clicking on OK, than you get back to the Molecules window:
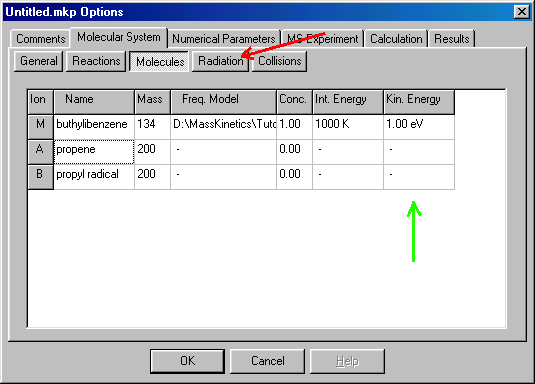
The last column in this dialog box (green arrow) is the initial kinetic energy (Kin. Energy), double clicking on it takes you to the Initial Kinetic Energy Distribution window: This is very similar to the previous one. For buthylbenzene you can specify fixed at 1 eV (red arrows) and accept it (OK). This takes you back to the Molecules window. You continue to the Radiation tab (red arrow):
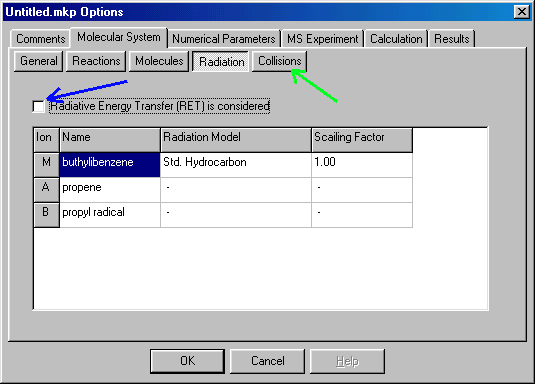
For the buthylbenzene example we do not consider radiative transitions, so leave the 'Radiative Energy Transfer (RET) is Considered' checkbox (blue arrow) unchecked, which is the default. (Note that it is not necessary to open/close the Radiation dialog box, but it is worth checking, if it is correctly set. Proceed to the Collisions tab (green arrow):
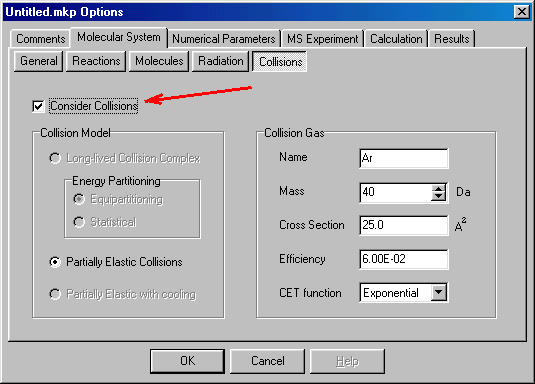
In the collision window you can define collision parameters. The checkbox (red arrow) should be checked (default setting) to consider collisional energy transfer. In the right hand side you can define the name and mass of the collision gas (click/type Ar and 40), the cross section of the collision (25) in square angstroms, efficiency (the fraction of the center of mass collision energy converted into internal energy (0.06, i.e. 6%), and the shape of CET function (collision energy transfer) – at present only exponential is available. You can proceed than to the next dialog box, Numerical Parameters:
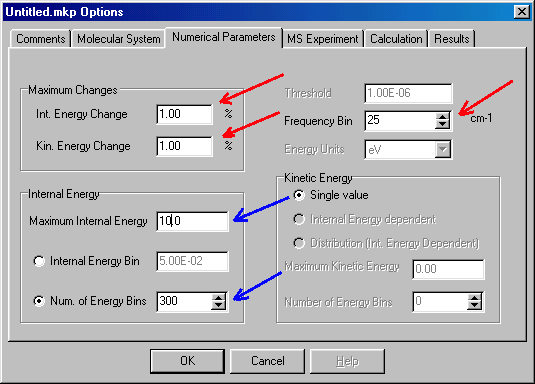
This box defines the numerical accuracy of the calculations. You have to experiment with the various tolerance limits yourself. You can change the pre-set values by click/type. In most cases you can leave the Int. Energy Change, Kin. Energy Change and Frequency Bin (red arrows) as they are (1%, 1% and 25 cm-1). You have to set the Maximum Internal Energy in eV units (to 10.0), and the Number of Energy Bins (to 300) by click/type (blue arrows). Note that the speed of calculations is inversely related to the square of the number of energy bins. After you finish with this dialog box, you can go to MS Experiment, which defines the mass spectrometric experiment you want to model:
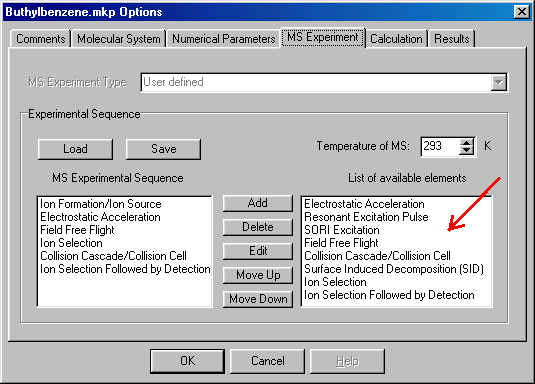
In the right hand side (red arrow) you have the possible experimental events (for a detailed description see L. Drahos and K. Vékey, J. Mass Spectrom. 36, 237 (2001) ), on the left (blue arrow) your experimental setup. You can select one item by clicking on it. If you select one on the right hand side you can move it to the left column by clicking the Add button. If you select one on the left, you can Delete, or Edit it or move it up or down the list (change the sequence. To edit an item you either click on Edit, or double click on the event.
EI ionization followed by CID in a typical triple quadrupole mass spectrometer can be defined using the following parameters: acceleration to 50 eV through 0.01 m distance, 0.50 m long distance to the collision region with selection of the molecular ion (1st quad), 0.20 m long collision cell (2nd quad), 0.50 m long distance through the 3rd quad to the detector. In the collision region the pressure of the collision gas is ~ 0.05 Pa (ca. 0.5 torr).
To set up such an experiment select/add events in the following sequence:
- ion formation (pre-selected)
- electrostatic acceleration
- field free flight (through the 1st quad)
- ion selection (of m/z 134)
- collision cascade/collision cell
- ion selection (through 3rd quad) followed by detection
When you have selected the proper experimental sequence, these will appear on the left hand side of this dialog box, as indicated in the figure above.
Now double-click on each item to define the specific parameters. Double-click on electrostatic acceleration, which takes you to the following dialog box:
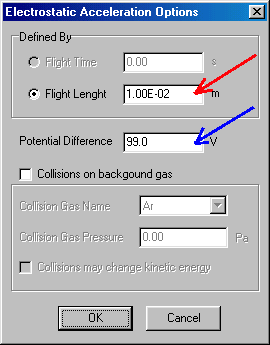
Here you should click on flight length, and specify 0.01 (m); red arrow. Specify the potential difference as 99 eV, (blue arrow) and finish by accepting it (OK), which brings you back to the MS Experiment dialog box. (Note that this defines kinetic (and therefore the lab-frame collision) energy to 100 eV.) Continue by double-clicking on field free flight (now representing the 1st quad):
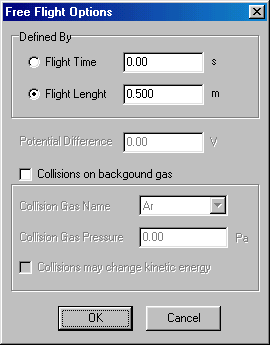
Now you click on flight length, and specify 0.5 (m), and accept it (OK). The next item is ion selection, double clicking brings you to:
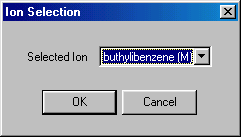
You select the Molecular ion ‘buthilbenzene (M)’, accept it (OK), than continue with collision cascade/collision cell:
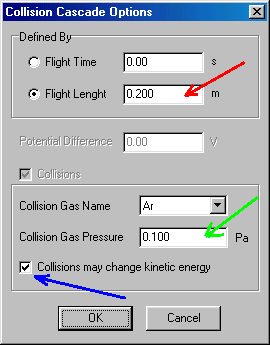
Here you select flight length, and specify 0.2 (m). Leave the Collisions box (red arrow) selected (you do want to study collisions). Specify the collision gas pressure (0.1, in Pa units, green arrow). Finally tick the Collisions may change the kinetic energy box (blue arrow). Note that this is the proper approximation, you may want to fix the kinetic energy in specific model calculations only. Finish by accepting these values (OK), than select the last item (ion selection followed by detection) by double clicking:
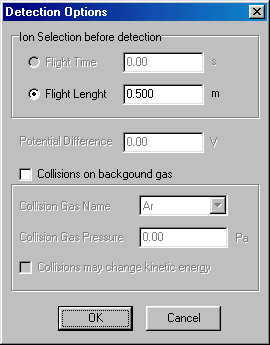
Here you have to check flight length, set it to 0.5 (m) and accept it (OK). Now you have finished defining your experiment, and you can continue with the Calculations tab (red arrow):
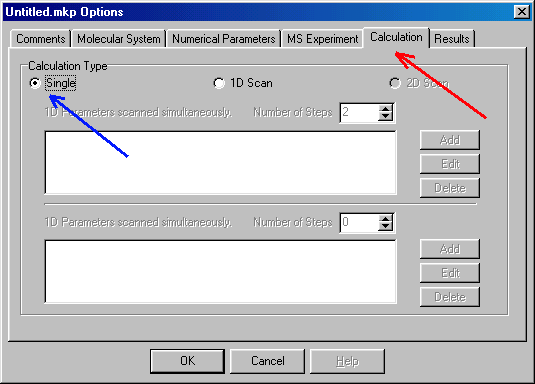
Here you specify single calculation (blue arrow), and you can switch to the Results tab (red arrow):
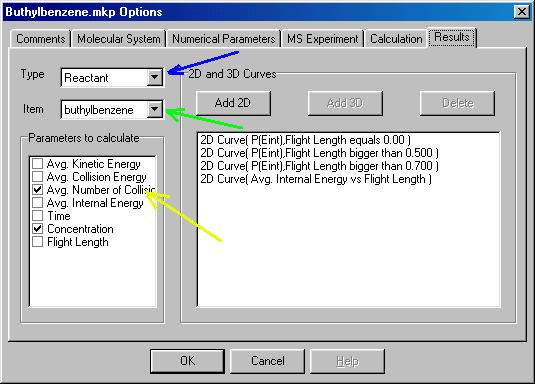
Here you can specify what sort of data you want to obtain for results. Let’s assume that you want to know the concentration of the molecular and fragment ions (relative to the initial amount you specified at Molecular System/Molecules/Conc.); the mean number of collisions; the effective temperature (as defined in the kinetic method) and the survival yield (molecular ion abundance relative to the sum of all ions). You also want to get the internal energy distribution of the molecular ion at the time of ion formation and before and after the collision cell. Finally, you want to know how does the mean internal energy change along the mass spectrometer.
In the Results dialog box you select Type to Reactant (blue arrow), Item to buthyilbenzene (green arrow) and check Avg. Number of Collisions and Concentration in the Parameters to Calculate box (yellow arrow). Next you set Item to propene (first fragment), and check Concentration in the Parameters to Calculate box:
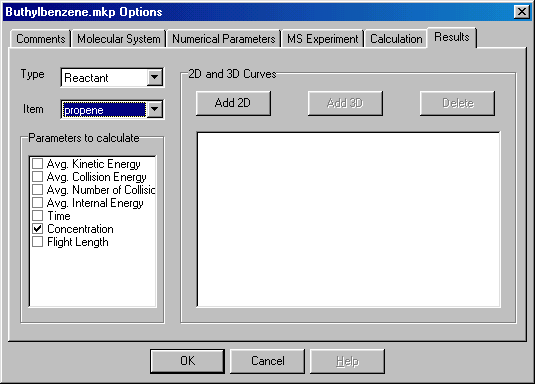
You continue in an analogous manner setting Item to propyl radical, and check Concentration in the Parameters to Calculate box again. Next you set Type to Molecular System (blue arrow), and check Effective Temperature and Survival Yield in the Parameters to Calculate box (red arrow):
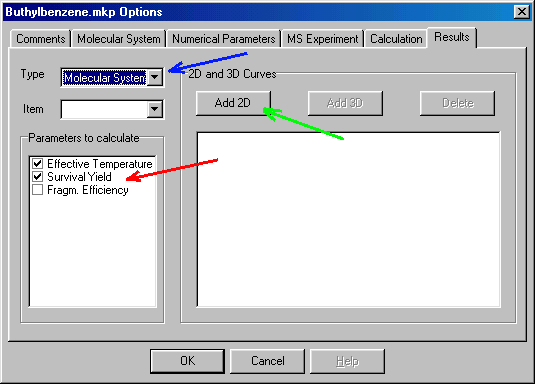
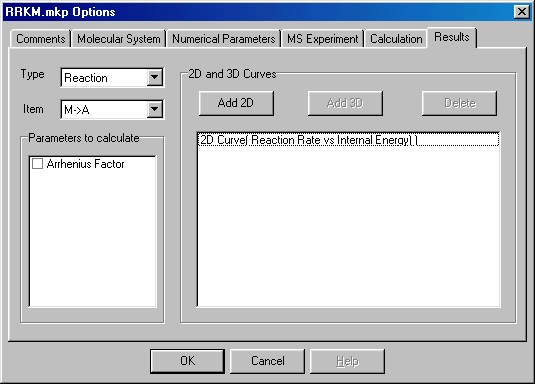
The next step is to ask for the internal energy distribution curves for the molecular ion. To do so, first set Type to Reactant, Item to buthylbenzene (Molecular Ion) and than click on the Add_2D tab (green arrow), which prompts you to fill out the following dialog box:
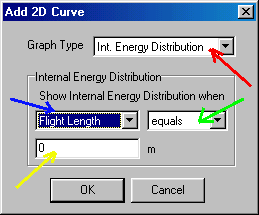
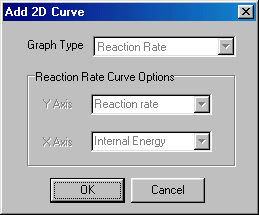
Here you set Graph Type to Int. Energy Distribution (red arrow), and set Show Internal Energy Distribution when to "Flight length" "equals", and click/type in "0" (blue, green and yellow arrows, respectively), than accept this selection by OK. You proceed in a similar manner to obtain the internal energy distribution immediately before the collision cell by clicking the Add_2D tab and setting again Graph Type to Int. Energy Distribution. Now you have to set Show Internal Energy Distribution when to "Flight Length" "bigger than" "0.5" – which distance corresponds to the beginning of the collision cell (measured from the ion source). Click Add_2D tab again, and set Graph Type to Int. Energy Distribution, and Show Internal Energy Distribution when to "Flight Length" "bigger than" "0.7" – which distance corresponds to the end of the collision cell (measured from the ion source). You are still in the Type = Reactant, Item = Molecular Ion case, and you click on the Add_2D tab again:
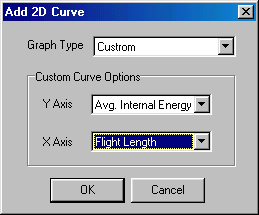
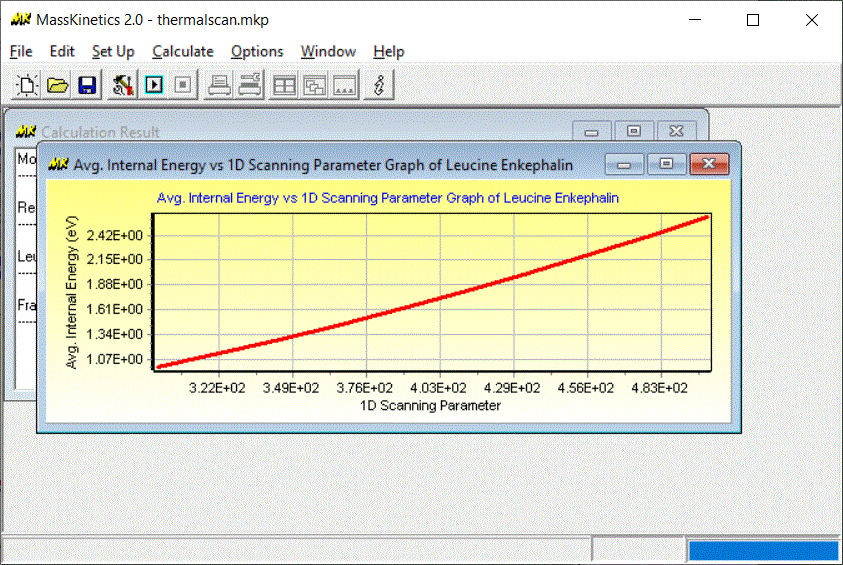
You set Graph Type to "Custom", y axis to "Avg. Internal Energy" and x axis to "Flight Length", and accept this choice by clicking OK. Now you have finished the Setup, so you should accept it by clicking OK at the bottom. This takes you back to the main window:
You should save it by clicking on the icon (blue arrow above) or by clicking on File/Save or File/Save as. Before you do so you may write comments in the Comments dialog box (which you can access from the Setup box).
Next you can prompt your computer to do the actual calculation, by clicking on Calculate (green arrow).
Calculation takes a few seconds only, you can monitor progress by the blue bar at the bottom right corner. When the calculations are finished, you will get the results in the main window:
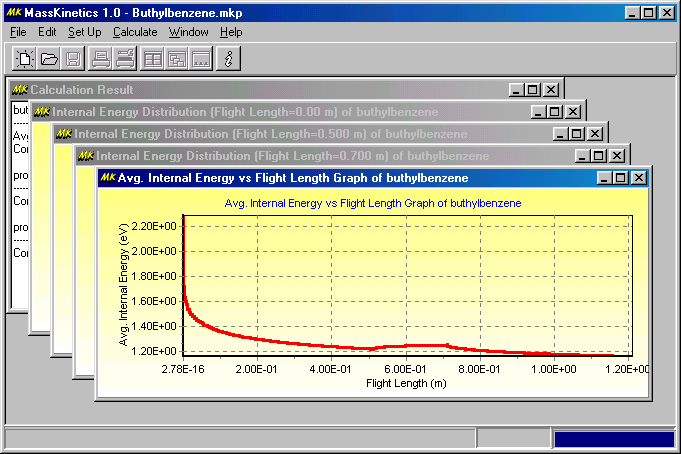
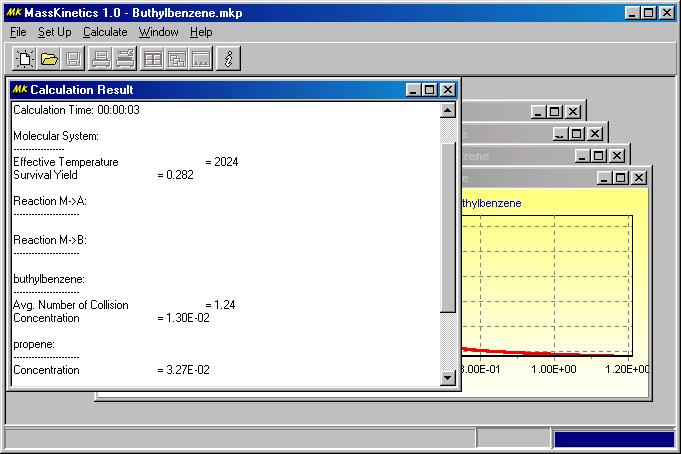
You can arrange them as in other Windows applications. The results show a fairly low survival yield (0.277) and a high effective temperature (2150 K), which is mainly the result of the high collision energy. There are only few collisions (1.24 on average). The concentrations of the molecular ion, propene loss and propyl radical loss are 1.34%, 3.42% and 0.0782%. These are all very small numbers – which indicates that most of the ions decompose either in the ion source or during flight through the 1st quadrupole. Those information can be found in the Calculation Result child window:
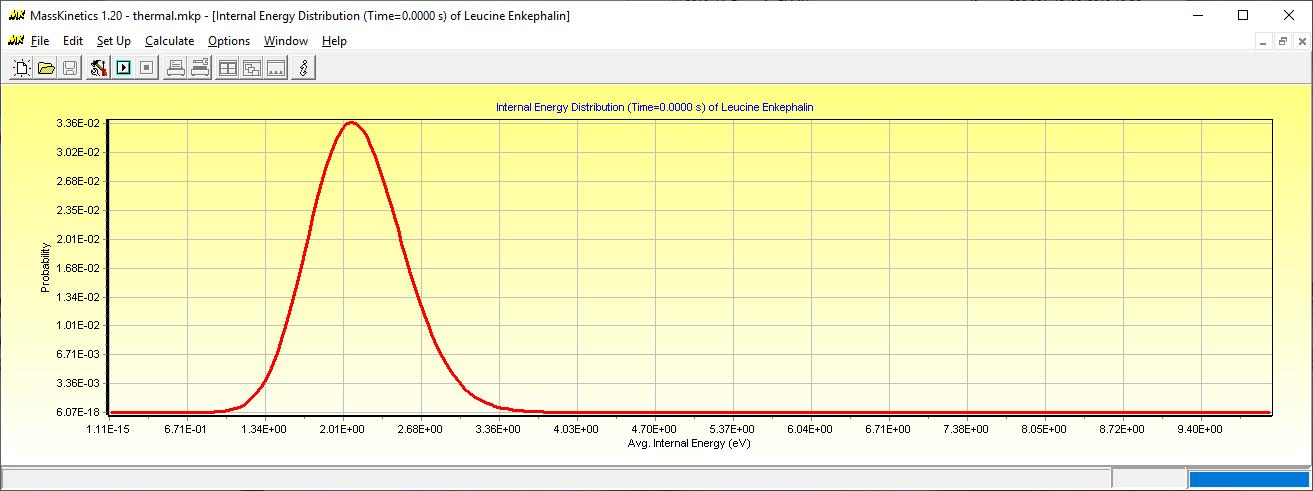
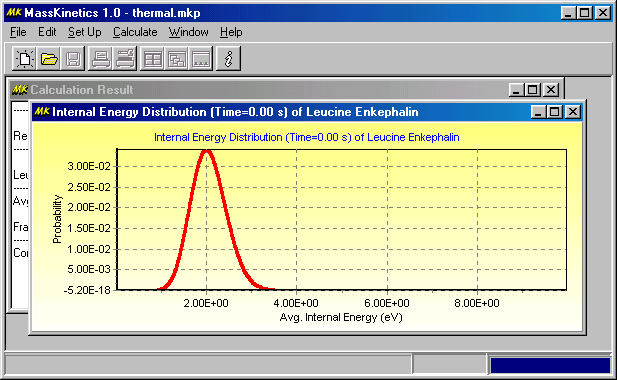
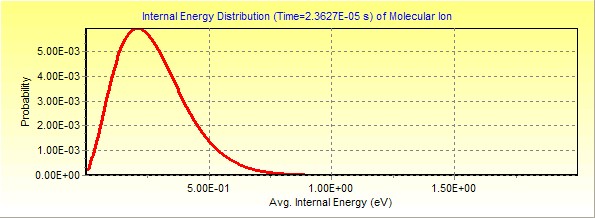
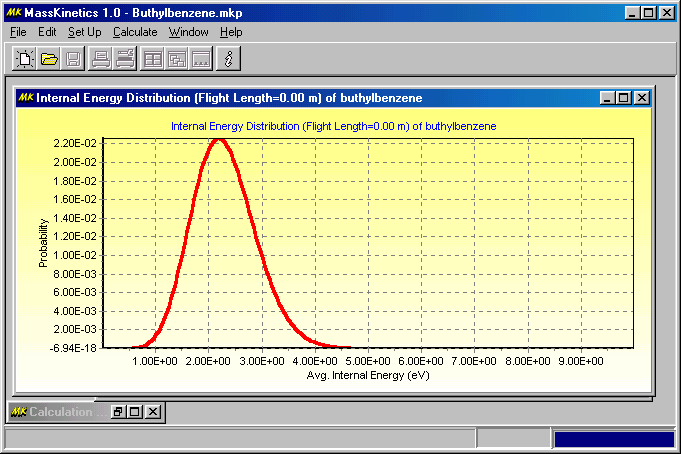
The first Figure shows the internal energy distribution of ions in the ion source (i.e. at 1000 K, as defined):
The next Figure shows the internal energy distribution of ions before the collision cell:
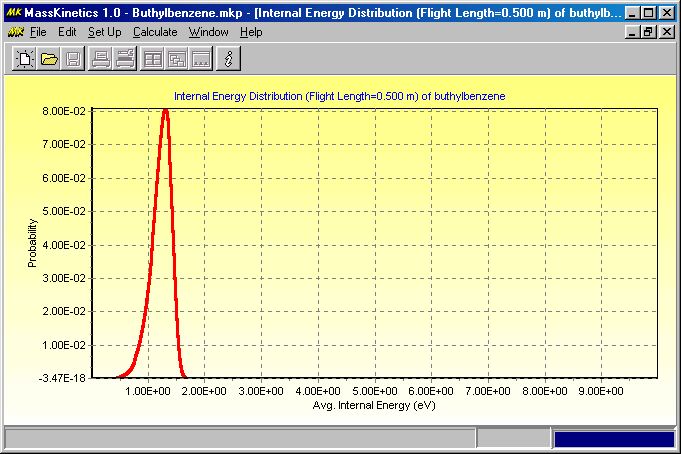
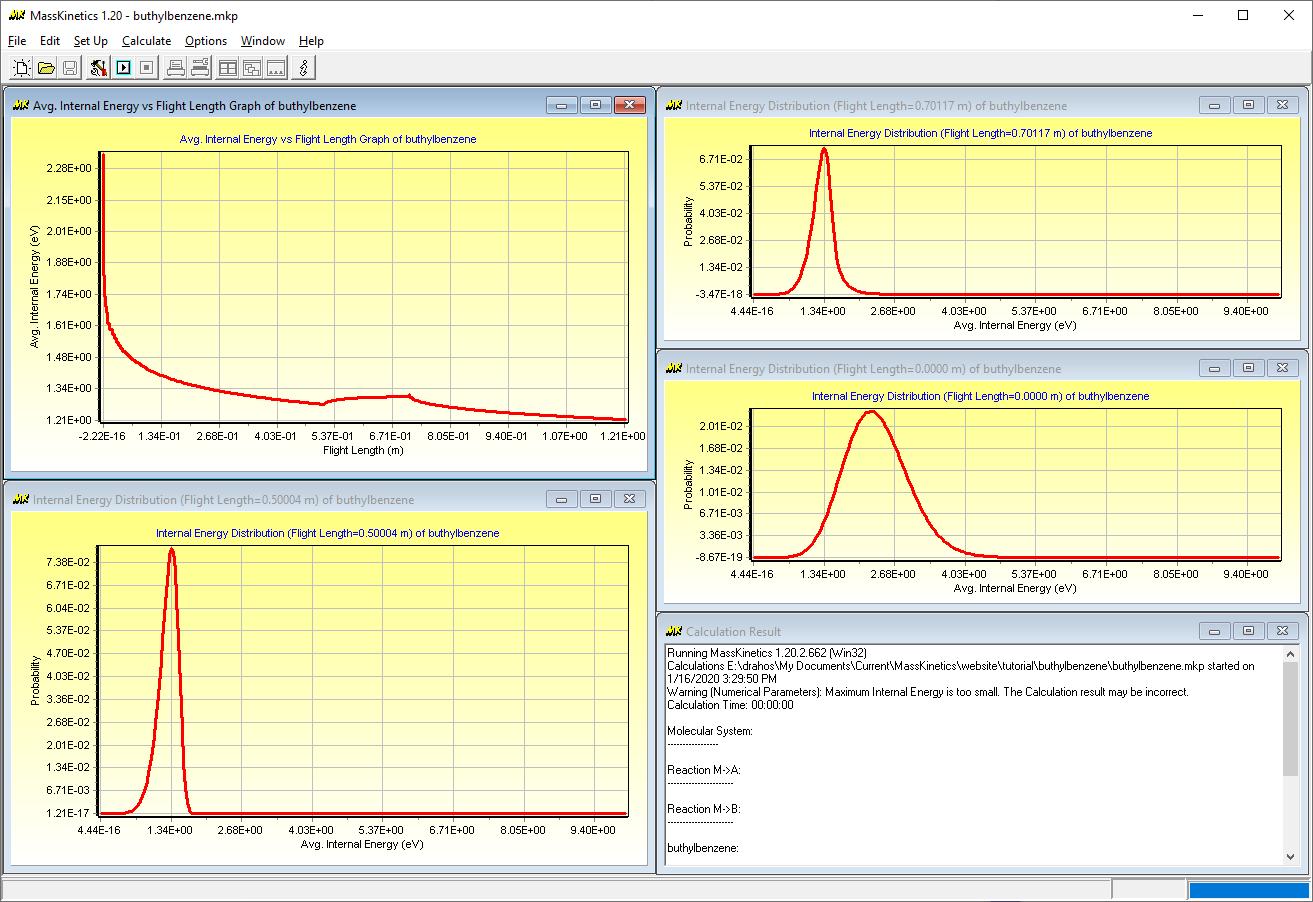
This indicates that most ions above ca. 1.5 eV internal energy decomposed during mass analysis (in accordance with the low ion yields) and only low internal energy ions enter the collision cell. The next Figure shows the internal energy distribution at the end of the collision cell:
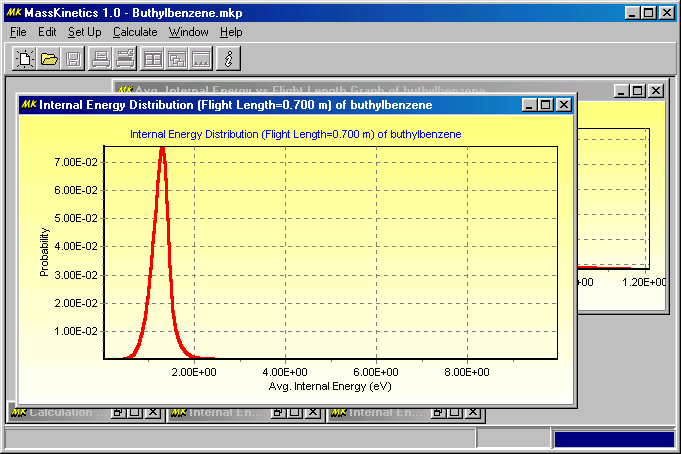
It shows a higher energy tail in the energy distribution curve, a consequence of collisional activation. (Note that ions which get to internal energy levels above ca. 2 eV decompose so fast, that they will not be apparent in this energy diagram). The last Figure shows the mean internal energy of the molecular ions as they move through the mass spectrometer:
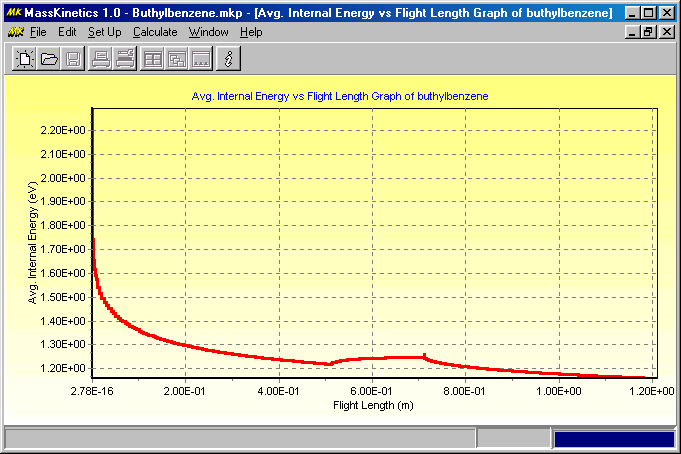
If you want to copy the diagrams you have to click Edit/Copy, this copies the current diagram onto the clipboard, and you can paste that into other programs (e.g. Word, Powerpoint, etc.) If you want to export the curve into text file, where you can manipulate them easily, click on the right mouse button over the graph and select Save Data as... menu item.
Click here to download this MassKinetics project file.